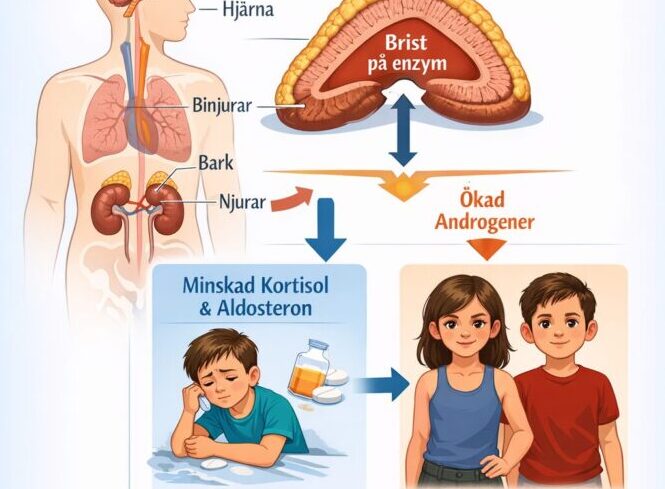
Kongenital binjurebarkshyperplasi betyder medfödd förstoring av binjurebarken, den del av binjurarna som normalt producerar hormonerna kortisol, aldosteron och vissa könshormoner. Vid CAH fungerar ett enzym, oftast 21‑hydroxylas, inte som det ska, vilket leder till brist på kortisol och ibland aldosteron samt överskott av manliga könshormoner (androgener).
Sjukdomen klassas som ett sällsynt hälsotillstånd och räknas också till gruppen DSD, “disorders/differences of sex development”, det vill säga tillstånd som påverkar könsutvecklingen. CAH kan därför ibland göra det svårt att avgöra kön vid födseln hos flickor, men påverkar inte de inre könsorganen.
Hur vanligt är CAH?
I Sverige beräknas svår 21‑hydroxylasbrist förekomma hos cirka 10 av 100 000 nyfödda, vilket motsvarar ungefär 10 barn med CAH per år. Hur vanliga de lindrigare formerna är i Sverige är oklart, eftersom de ofta upptäcks senare i livet. CAH räknas som en sällsynt diagnos, men är en av de vanligare medfödda hormonrubbningarna som upptäcks via nyföddhetsscreeningen.
Internationellt anges förekomsten av klassisk (svår) CAH ofta till cirka 1 på 13 000–15 000 nyfödda, med variation mellan olika länder och befolkningsgrupper.
Symtom vid CAH
Vanliga symtom vid svår (klassisk) CAH
- Tecken på saltbrist hos spädbarn: dålig viktuppgång, kraftiga kräkningar, uttorkning, slöhet och ibland chocktillstånd under de första levnadsveckorna.
- Lågt natrium (salt) och högt kalium i blodet, vilket visar på allvarlig rubbning av saltbalansen.
- Kortisolbrist, som kan ge ökad känslighet för infektioner, lågt blodsocker och svårigheter att klara fysisk och psykisk stress.
Hos flickor kan de yttre könsorganen bli mer eller mindre “viriliserade”, vilket betyder att de utvecklas åt manligt håll: förstoring av klitoris och sammanväxning av blygdläppar, medan livmoder och äggstockar är normala. Hos pojkar är yttre könsorgan normalt manliga, vilket gör att svår CAH ibland upptäcks först när saltbristen ger akuta symtom.
Symtom vid lindrigare och icke‑klassiska former av CAH
- Ingen eller mycket liten symtomgivande påverkan vid födseln.
- Tidig tillväxtspurt och tecken på “för tidig pubertet”, som tidig kroppsbehåring, akne och vuxen svettlukt redan i förskoleåldern.
- Hos ungdomar och vuxna kvinnor kan symtomen vara oregelbunden eller utebliven mens, ökad kroppsbehåring, akne och ibland nedsatt fertilitet.
Alla med CAH, oavsett svårighetsgrad och kön, kan få symtom på kortisolbrist i samband med infektioner, operationer, olyckor eller annan kraftig stress, till exempel feber eller större fysisk ansträngning. Vuxna med svårare former löper också ökad risk att utveckla godartade tumörer i binjurarna eller testiklarna som kan påverka fertiliteten.
Orsak och ärftlighet vid CAH
Den vanligaste formen av CAH beror på mutationer (förändringar) i genen CYP21A2 på kromosom 6, som är ritningen för enzymet 21‑hydroxylas. Enzymet behövs för att bilda kortisol och aldosteron, och när det inte fungerar som det ska, minskar nivåerna av dessa hormoner samtidigt som förstadierna styrs om till manliga könshormoner.
Kortisol är ett livsviktigt stresshormon som bland annat hjälper till att hålla blodtryck och blodsocker på en stabil nivå. Aldosteron är ett hormon som reglerar kroppens salt- och vätskebalans, och brist på aldosteron kan ge livshotande saltförlust hos nyfödda. Överskottet av manliga hormoner under fosterlivet gör att flickors yttre könsorgan kan utvecklas åt manligt håll, medan pojkar får förhöjda nivåer först senare.
CAH nedärvs autosomalt recessivt, vilket betyder att:
- båda föräldrarna vanligtvis är friska bärare av en förändrad gen,
- vid varje graviditet är risken 25 procent att barnet ärver två förändrade gener och får sjukdomen,
- det är 50 procent risk att barnet blir frisk bärare, och 25 procent chans att barnet varken får sjukdomen eller bär på genförändringen.
Om en person med CAH får barn med någon som inte är bärare blir alla barn friska bärare, men får inte sjukdomen. Om en person med CAH får barn med en bärare är risken 50 procent att barnet får sjukdomen.
Diagnos och utredning vid CAH
Sedan 1986 ingår CAH i den svenska nyföddhetsscreeningen (PKU‑provet), där ett blodprov tas efter 48 timmars ålder. Vid 21‑hydroxylasbrist är halten av hormonet 17‑hydroxyprogesteron (17‑OHP) förhöjd, vilket gör att barn med svårare form kan upptäckas innan de blir allvarligt sjuka.
Utredningen fortsätter oftast med blod- och urinprov för att mäta hormonnivåer och DNA‑analys som kan påvisa mutationer i CYP21A2 och bekräfta diagnosen.
Alla lindrigare former av CAH upptäcks inte i PKU‑provet, utan kan visa sig först genom tidig pubertetsutveckling, avvikande tillväxt eller fertilitetsproblem. I familjer där könet är svårt att fastställa vid födseln ska utredningen påbörjas omgående hos specialistteam med erfarenhet av DSD, för att både medicinska och psykologiska behov ska tas om hand.
Genetisk vägledning erbjuds i samband med diagnos, där familjen får information om sjukdomen, ärftlighet, risker för framtida barn och möjligheter till anlagsbärar- och fosterdiagnostik, inklusive preimplantatorisk genetisk diagnostik (PGD) vid IVF.
Behandling och stöd vid CAH
Läkemedelsbehandling
Behandlingen vid svårare CAH är livslång och består framför allt av:
- Glukokortikoider (kortison, oftast hydrokortison) för att ersätta bristen på kortisol och dämpa den överdrivna produktionen av androgener.
- Mineralokortikoider (till exempel fludrokortison) för att ersätta bristen på aldosteron och stabilisera saltbalansen.
Doserna justeras efter barnets ålder, vikt, tillväxt, hormonnivåer och vardagliga aktiviteter, och följs regelbundet av barnendokrinolog. Under de första levnadsåren kan extra salt behöva ges.
Vid feber, infektioner, olyckor eller operationer behöver kortisondoserna höjas till så kallade stressdoser, eftersom kroppen då normalt skulle producera mer kortisol. Vid magsjuka kan det behövas kortison som injektion, och familjer får därför instruktioner om hur de ska agera vid akuta situationer.
Kirurgi och etiska överväganden
För flickor med kraftigt viriliserade yttre könsorgan kan kirurgi ibland bli aktuellt, men synen på när och hur detta ska göras har förändrats. Idag betonas vikten av att ingreppen görs så skonsamt som möjligt och, om det är möjligt, vid en tidpunkt då personen själv kan vara delaktig i besluten.
Psykologiskt och psykosocialt stöd
Familjen erbjuds tidigt kontakt med psykolog och kurator, både för att hantera den första krisen och för att få långsiktigt stöd. Barnet behöver fortlöpande åldersanpassad information om diagnosen och sin behandling. Behovet av stöd ökar ofta i puberteten och i samband med frågor om identitet, sexualitet och framtida fertilitet.
För vuxna med CAH är regelbunden kontakt med endokrinolog och ibland gynekolog eller urolog viktigt, liksom möjlighet till psykologiskt stöd vid behov.
Botande behandling i form av genterapi finns idag inte, men med optimal hormonbehandling, uppföljning och stöd kan många leva ett aktivt liv med god livskvalitet.
Att leva med sjukdomen CAH
CAH påverkar ofta vardagen, men påverkan ser olika ut beroende på ålder, sjukdomens svårighetsgrad och hur väl behandlingen är inställd. För små barn kan täta kontroller, medicinering flera gånger om dagen och oro för infektioner ta mycket energi, både för barnet och föräldrarna.
I förskola och skola kan det behövas information till personalen om sjukdomen, särskilt kring behovet av medicin, extra kortison vid feber och vad som är tecken på akut försämring. Under tonåren kan frågor om kroppen, könsutveckling, sexualitet och fertilitet bli extra känsliga, vilket gör kontinuerligt stöd från vården och möjlighet att träffa andra i samma situation viktigt.
Många vuxna med CAH arbetar, bildar familj och lever ett aktivt liv, men kan behöva regelbunden kontroll av blodtryck, vikt, skelett, fertilitet och psykisk hälsa. Ett viktigt praktiskt råd är att alltid bära ett kort eller halsband som visar diagnosen och behovet av kortison vid akuta situationer, samt att ha med sig extra mediciner vid resor.
I Sverige finns både specialiserade expertteam för CAH och DSD, Centrum för sällsynta diagnoser (CSD) vid universitetssjukhusen samt patientföreningar som erbjuder erfarenhetsutbyte, information och gemenskap.
Prognos vid CAH
Med tidig diagnos, korrekt doserad hormonbehandling och regelbunden uppföljning är prognosen vid CAH generellt god. De flesta kan växa upp, gå i skolan, arbeta och ha familj, även om sjukdomen kräver livslång behandling och egenvård.
Vid svår, saltförlorande form är risken för allvarliga komplikationer störst om diagnosen missas eller behandlingen blir otillräcklig, särskilt under de första levnadsåren. Vid lindrigare former kan prognosen vara mycket god, men det finns ökad risk för bland annat fertilitetsproblem och vissa metabola komplikationer om hormonnivåerna inte är välkontrollerade.
Biverkningar av långvarig kortisonbehandling, som kortvuxenhet, övervikt, benskörhet och försämrad ämnesomsättning, har minskat i takt med förbättrade behandlingsstrategier. Fortsatt forskning och nationella kvalitetsregister bidrar till att finjustera behandlingen och förbättra långtidsprognosen ytterligare.
Forskning och framtid
Molekylärgenetiken bakom CAH är idag väl kartlagd, och kopplingen mellan olika mutationer i CYP21A2 och sjukdomens svårighetsgrad är relativt tydlig. Neonatalscreening har förbättrat tidig upptäckt och gett bättre kunskap om hur vanligt tillståndet är i Sverige.
Aktuell forskning fokuserar bland annat på:
- finjustering av glukokortikoidbehandling för att minimera biverkningar och optimera tillväxt och livskvalitet,
- nya läkemedelsformer, som mer långverkande kortisonpreparat och beredningar som bättre efterliknar kroppens naturliga dygnsrytm,
- förbättrade metoder för fertilitetsbehandling och stöd till vuxna med CAH.
Genterapi diskuteras som en möjlig framtida behandling, men bedöms fortfarande ligga långt fram i tiden och är ännu inte en del av klinisk vardag.
Vanliga frågor (FAQ)
Är CAH smittsamt?
Nej, kongenital binjurebarkshyperplasi är inte smittsam utan beror på ärftliga genförändringar som förs vidare via arv.
Kan syskon också drabbas?
Ja, eftersom sjukdomen är autosomalt recessivt ärftlig finns det vid varje graviditet med samma föräldrar 25 procents risk att barnet får CAH, 50 procents risk att bli bärare och 25 procents chans att varken få sjukdom eller bärare.
Finns det behandling?
Ja, det finns effektiv behandling med hormoner i tablettform som ersätter bristen på kortisol och aldosteron, och behandlingen är oftast livslång. Vid rätt dosering kan många leva ett aktivt liv med god livskvalitet.
Kan man få egna barn om man har CAH?
Många med CAH kan få barn, även om fertiliteten kan vara nedsatt och ibland kräver utredning och behandling hos specialist. Det är också viktigt med genetisk vägledning kring risker och möjligheter i samband med graviditet.
Behöver barnet särskild uppmärksamhet vid infektioner eller operationer?
Ja, vid feber, infektioner, olyckor eller operationer behöver kortisondoserna höjas till stressdoser för att kroppen ska klara belastningen. Familjen får detaljerade instruktioner och ibland injektionsläkemedel att använda vid akuta situationer.
Var finns mer information och stöd kring CAH?
- Socialstyrelsens kunskapsdatabas om sällsynta hälsotillstånd – detaljerad medicinsk information om kongenital binjurebarkshyperplasi, inklusive behandling, resurser och litteratur.
- NIH/GARD – Congenital adrenal hyperplasia – engelskspråkig information om CAH med fokus på internationella data, genetik och symtom.
- Centrum för sällsynta diagnoser (CSD) – finns vid alla universitetssjukhus och erbjuder vägledning, information och samordning kring sällsynta diagnoser.
- Expertteam för DSD/CAH vid universitetssjukhusen – särskilt team med barnendokrinologer, kirurger, psykologer med flera som har erfarenhet av CAH och andra tillstånd som påverkar könsutvecklingen.
Patient- och intresseorganisationer för CAH
- Riksföreningen för congenital adrenal hyperplasia (CAH) – svensk patientförening för barn och vuxna med CAH och deras familjer, erbjuder information, träffar och erfarenhetsutbyte
- Riksförbundet Sällsynta diagnoser – paraplyorganisation för personer med sällsynta diagnoser i Sverige
Det finns även stöd från Ågrenska, som arrangerar vistelser och kursdagar för familjer och vuxna med sällsynta diagnoser, samt sociala medier-grupper där man kan dela erfarenheter med andra i liknande situation.